El genoma humà es pot entendre de manera simplificada com una llarga cadena d’àcid desoxiribonucleic (ADN) formada per quatre tipus de nucleòtids (A=adenina; C=citosina; G=guanina; T=timina). Una fracció minoritària del genoma és transcriu en cadenes d’àcid ribonucleic (ARN) formada per quatre tipus de nucleòtids (A, C, G; U=uracil). I una part d’aquest transcriptoma es tradueix a proteïna, és a dir a cadenes polipeptídiques de 20 tipus d’aminoàcids. La traducció implica convertir triplets de nucleòtids d’ARN (codons) en aminoàcids, cosa que fa l’aparell ribosòmic d’acord amb el codi genètic definit per les ARN-aminoacil-transferases. Hom pot definir, doncs, una genoma humà de consens integrat aproximadament per 3100 milions de parells de nucleòtids. D’aquestes posicions, però, n’hi ha 600 milions que són polimòrfiques, en el sentit que hi ha almenys una variant amb una freqüència gènica de l’1%. El nombre encara augmenta si hi sumem les variants més rares. Tan sols una fracció de les variants del genoma té conseqüències en el proteoma. Quan una variant gènica comporta un canvi en la seqüència d’aminoàcids d’una proteïna diem que és una ‘variant de canvi de sentit’. Les variants de canvi de sentit són al darrera d’un terç de les malalties genètiques humanes, i se n’han descrit desenes milions de variants de canvi de sentit. De la immensa majoria se’n desconeixen les conseqüències funcionals. El laboratori de Ben Lehner, del Centre de Regulació Genòmica de Barcelona, ha treballat en una anàlisi experimental massiva de variants de canvi de sentit sobre dominis de proteïnes humanes. En un article a la revista Nature, amb Antoni Beltran com a primer autor, presenten la quantificació de l’efecte de més de 500.000 variants sobre l’abundància de més de 500 dominis de proteïnes humans. Ho han fet a través de tècniques de síntesi d’ADN i d’experiments de selecció cel·lular en llevat. De les variants de canvi de sentit descrites en la bibliografia com a patogèniques, el 60% comporten una reducció de l’estabilitat de la proteïna afectada. Val a dir que aquesta proporció segons cada proteïna i cada malaltia genètica, i és especialment elevada quan es tracta de malalties genètiques recessives. En aquestes malalties recessives, cal que la mutació afecti les dues còpies del genoma diploide perquè s’hi manifesti la patologia. Beltran et al. combinen mesures d’estabilitat amb models de llenguatge proteic per anotar posicions funcionals del proteoma humà. Els efectes de les mutacions sobre l’estabilitat són força conservats en dominis proteics homòlegs, la qual cosa assenyalen Beltran et al. que facilita la predicció de l’estabilitat de famílies senceres de proteïnes a través de models energètics. La base de dades generada pot ajudar a la interpretació clínica de variants gèniques, i també pot ajudar en el desenvolupament de mètodes computacionals sobre les seves conseqüències funcionals.
Beltran et al. han conduït experiments de mutagènesi sobre el dominioma humà.
El dominioma humà
Les genoteques o llibreries foren dissenyades per Antoni Beltran, qui realitzà tots els experiments i anàlisi. Xiang’er Jiang i Yue Shen, de BGI Research, de Changzhou sintetitzaren les genoteques de variants. El projecte havia estat concebut per Beltran i Ben Lehner, i foren ells dos els qui dissenyaren les anàlisi i redactaren l’article.
Aquesta recerca compta amb finançament de la Unió Europea, del ministeri espanyol de Ciència i Innovació, de la Bettencourt Schueller Foundation, de la Generalitat de Catalunya, de l’EMBO, de la Fundació Nacional de Ciència Natural de la Xina i de la Província de Jiangsu. Els autors agraeixen les contribucions de tots els membres del laboratori Lehner.
Lehner és l’autor corresponsal. Declara que és fundador i accionista d’ALLOX, spin-off dedicada al disseny de nous fàrmacs dirigits a llocs al·lostèrics de proteïnes. Lehner trameté l’article a la revista Nature el 26 d’abril del 2024. L’article fou revisat per Willow Coyote-Maestas, Jacob Kitzman i un altre revisor. El 8 de novembre l’article fou acceptat, i es publicà el 8 de gener del 2025.
El genoma humà codifica un proteoma humà integrat per més de 20.000 proteïnes. El catàleg OMIM (On-line Mendelian Inheritance in Man) és hereu del llibre de Victor McKusick Mendelian Inheritance in Man, que en el 1966, enumerava 1400 fenotips d’herència mendeliana. Actualment el nombre d’entrades és de 27631 fenotips. Entre aquestes entrades OMIM enumera variants de canvi de sentit de gairebé 5000 proteïnes que causen malalties d’herència mendeliana. Però el nombre de variants de canvi de sentit de les que hom no coneix les conseqüències funcionals és enorme.
En la població humana general, de 8.025 milions de persones, hi ha prou espai com perquè la majoria de variants compatibles amb la vida hi siguin presents, si més no, en alguna d’aquestes persones. Un projecte com UK Biobank ha aconseguit la seqüenciació completa del genoma de 500.000 voluntaris. Els esforços experimentals han quantificat els efectes de variants en un petit nombre de proteïnes. Per a la resta hom confia en el desenvolupament de predictors computacionals d’efectes de variants (VEPs), que ara per ara no són capaços de distingir entre variants patogèniques o benignes. Aquesta distinció passa per identificar els mecanismes moleculars que fan que una variant gènica provoqui una malaltia. Alhora, la patologia molecular d’una variant gènica és ben rellevant per al desenvolupament de teràpies i el disseny d’assaigs clínics.
Entre els possibles mecanismes patogènics de les variants gèniques o mutacions hi hauria la desestabilització de la proteïna afectada, que comportaria una reducció de la quantitat d’aquesta proteïna en els tipus cel·lulars que l’expressen. Altres mecanismes afectarien interaccions moleculars especifiquen, fent que la proteïna perdés funcionalitats necessàries o guanyés funcionalitats perjudicials.
En l’estudi de la funcionalitat de proteïnes resulta essencial el concepte de domini. Un domini proteic és una unitat estructural de plegament independent. La majoria de proteïnes contenen múltiples dominis.
En el genoma humà hom ha descrit, per exemple, més de 200 homeodominis que tenen la capacitat d’unir-se específicament a l’ADN i de controlar l’expressió gènica. També s’han descrit més de 250 dominis PDZ que fan de mitjancers en interaccions proteïna-proteïna. El dominis proteics consten d’una mediana de 100 aminoàcids. Això, i el fet que tinguin un plegament independent, fan que la dominiòmica sigui una bona estratègia en la caracterització a gran escala de variants de canvi de sentit.
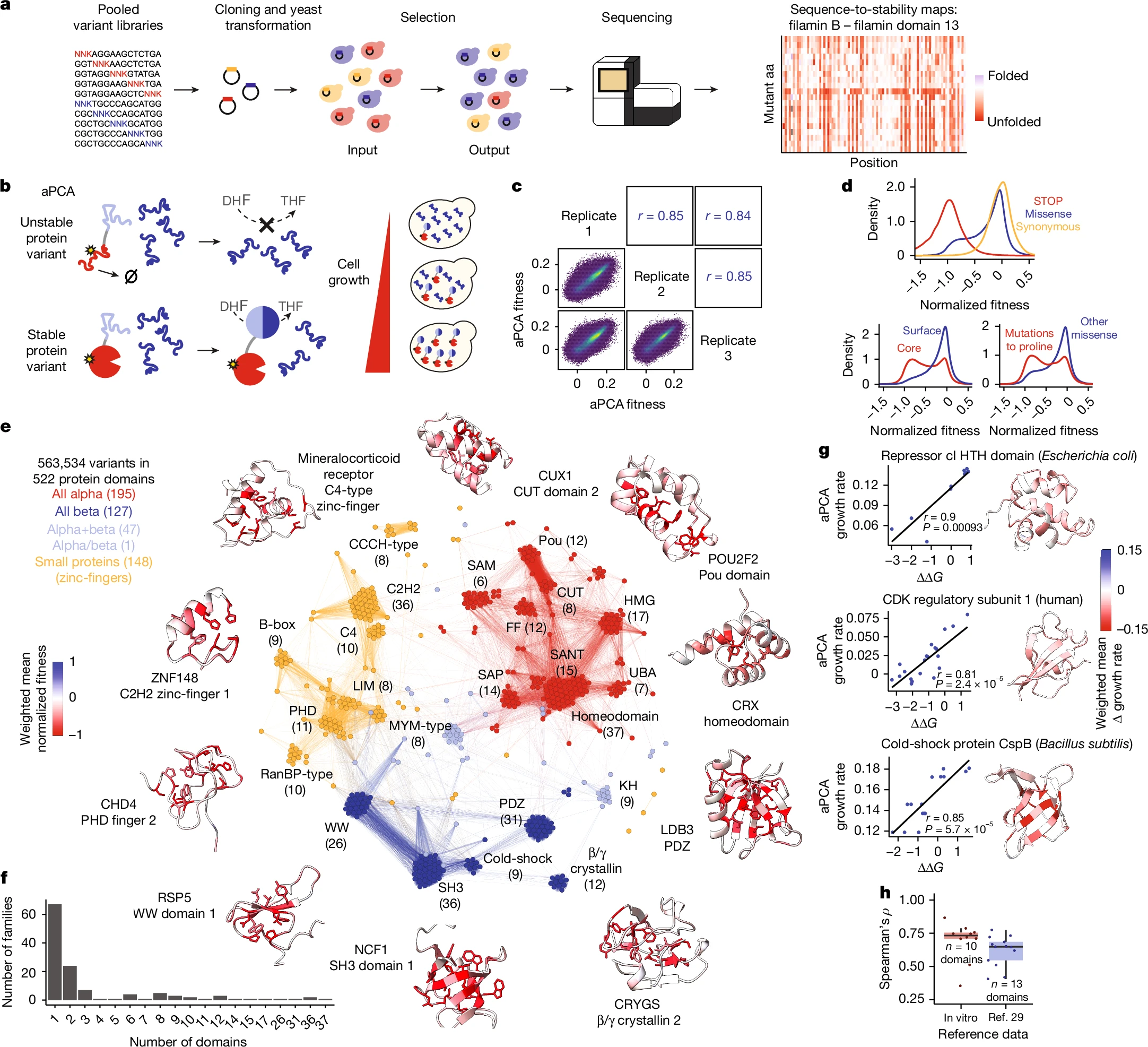
Beltran et al. utilitzen un assaig altament validat per quantificar els efectes de variants sobre l’abundància de proteïnes en cèl·lules. Aquestes variants són produïdes mitjançant una mutagènesi a gran escala de dominis proteics humans. Han construït la base de dades ‘Human Domainome 1’, que reporta l’efecte de més de 500.000 variants de canvi de sentit sobre l’estabilitat de més de 500 dominis proteics humans.
Mutagènesi de saturació de lloc de dominis proteics humans
El punt de partida foren els dominis anotats de proteïnes intracel·lulars humanes, és a dir excloent les extracel·lulars o secretades, i proteïnes transmembrana.
Beltran et al. han aplicat tecnologia mMPS (síntesi paral·lela massiva en microxips) per construir una biblioteca de 1.230.584 variants aminoacídiques de 1.248 dominis proteics. Es tracta d’una mutagènesi de saturació, ja que cada posició aminoacídica ha estat mutada a tots i cadascun dels altres 19 aminoàcids possibles.
La qualitat de la biblioteca ha estat examinada per seqüenciació, amb un grau de cobertura del 91%.
Les seqüències foren optimitzades segons la taula d’ús de codons de Saccharomyces cerevisiae, bo i evitant el codó GCT d’alanina, ja que es relaciona amb els llocs de tall de l’enzim de restricció HindIII. Les llibreries eren clonades en el plàsmid pGJJ162.
L’efecte d’aquestes variants sobre l’estabilitat del domini proteic ha estat quantificada en un assaig d’abundància aPCA (complementació de fragment de proteïna) en un sistema de selecció intracel·lular de S. cerevisiae BY4741. En aquest aPCA, el domini proteic d’interès és expressat com a fusió amb un fragment d’un enzim essencial. La concentració d’aquest enzim essencial determina linealment la taxa de creixement cel·lular al llarg d’un mínim de tres ordres de magnitud. Els efectes de les variants sobre l’abundància de la proteïna de fusió es quantifiquen amb una seqüenciació massiva de manera que hom mesura el canvi en les freqüències de variant entre les poblacions cel·lulars d’entrada i sortida en experiments de selecció. En un sol experiment es fa la clonació, transformació i selecció de centenars de variants de diverses proteïnes.
En total, Beltran et al. realitzaren 27 experiments de transformació, selecció i seqüenciació. Els 27 experiments es corresponen a triplicats biològicament independents de 9 sub-llibreries. Després del procés de filtració construeixen una base de dades de mesures d’abundància cel·lular per a 563.534 variants de 522 dominis proteics, dels quals 503 són de proteïnes humanes.
Els triplicats indiquen que les mesures d’abundància són altament reproduïbles (r=0,85). També ofereixen una bona correlació amb mesures in vitro independents d’estabilitat de plegament de proteïna fetes sobre 10 dominis. Finalment, també tenen una bona correlació amb mesures massives d’estabilitat fetes sobre 13 dominis amb assaigs de sensibilitat a proteasa.
Els 522 dominis que abasta l’estudi són estructuralment diversos. N’hi ha 195 de la classe estructural tot-alfa; 127 de la classe estructural tot-beta; 48 de classe estructural mixta alfa-beta; i 148 de la classe estructura de dit de zinc d’unió a metall. Això es correspon a 127 famílies diferents de dominis, incloent-hi 14 famílies amb 10 o més dominis homòlegs i 97 famílies amb 1 o 2 dominis homòlegs. Encara queda espai per recórrer, ja que representen únicament el 2,1% de totes les proteïnes humanes, l’1,2% de tots els dominis i el 2,0% de totes les famílies de dominis.
D’aquests 522 dominis, n’hi ha 275 codificats per gens implicats en malalties humanes. I n’hi ha 108 dominis que contenen variants patogèniques prèviament anotades.
En termes generals, les mutacions en els nuclis interns dels dominis són més perjudicials que les mutacions en les zones superficials. Les mutacions que introdueixen aminoàcids polars tenen un efecte desestabilitzador més fort en les nuclis dels dominis. Inversament, les mutacions que introdueixen aminoàcids hidròfobs o apolars tenen un efecte desestabilitzador més fort en les superfícies.
Les mutacions que introdueixen prolines tenen un efecte desestabilitzador tant en els nuclis com les superfícies dels dominis. Ara bé, l’efecte desestabilitzador de la prolina és més accentuat en les làmines beta i en les hèlixs que no pas en els girs.
Una avaluació dels predictors EVPs
Beltran et al. recorden que la seva base de dades multiplica per cinc les mesures d’estabilitat de mutacions en proteïnes humanes fins ara realitzades. Per això gosen aplicar aquestes dades a avaluar els predictors computacionals EVPs.
Els EVPs prediuen l’estabilitat d’una proteïna a través de la seva seqüència aminoacídica. Beltran et al. destaquen la predicció raonable que ofereix el model de llenguatge proteic ESM1v o el model generatiu profund EVE. El millor dels predictors d’estabilitat seria la xarxa neural gràfica ThermoMPNN.
Allà on més fallen aquests models és en els dominis petit de dits de zinc. Això podria explicar-se pel que aquests dominis requereixen un grup prostètic, concretament un metall, per arribar a la plena estabilitat.
El rol evolutiu de l’estabilitat de plegament
L’estabilitat de plegament és una de les moltes propietats biofísiques que contribueixen a la conservació evolutiva de funcions i seqüències proteiques. No obstant, funcions proteiques com la unió a altres proteïnes, a àcids nucleics, a metabòlits, o com la catàlisi de reaccions bioquímiques, requereixen una certa flexibilitat de plegament.
Beltran et al. han comparat l’estabilitat quantificada en els seus experiments amb els paràmetres d’aptitud evolutiva calculats per ESM1v per a un total de més de 500.000 variants de més de 500 dominis. L’estabilitat de proteïna explicaria una mediana del 30% de la variança d’aptitud. Aquest valor puja al 40% quan es tracta de dominis tot-beta, i baixa al 25% en dominis tot-alfa i mixtos. Això s’explica pel que les hèlixs alfa tenen una major tolerància estructural a les mutacions respecte les làmines beta.
L’estabilitat és relativament important per a l’aptitud evolutiva de dominis com SH3, WW, dit PHD i interaccions proteïna-proteïna Pointed. No ho és gaire per als dominis HMG-box d’unió a ADN, o per als dominis de dit de zinc C4 del tipus d’hormones nuclears, o per als homeodominis, possiblement perquè en aquests casos les interaccions amb l’ADN són crucials.
Els llocs funcionals de les proteïnes
En principi mutacions en llocs d’unió, en centres actius i en llocs de control al·lostèric tenen un efecte funcional superior a l’atribuïble exclusivament a un canvi en l’estabilitat proteica.
Beltran et al. exploren aquesta relació amb una comparació amb la predicció d’aptitud evolutiva d’ESM1v. Per a dominis individuals no hi ha una relació lineal entre l’abundància experimental de la proteïna i la predicció computacional d’aptitud. Per amb corbes sigmoidals és possible modelitzar aquesta relació per a 426 dominis, i identificar mutacions amb efectes sobre l’aptitud no atribuïbles a l’estabilitat. Així identifiquen un total de 102.231 mutacions que tenen un efecte funcional no associat a l’estabilitat proteica: són el 24% del total. Aquestes mutacions assoleixen una major proporció en llocs funcionals anotats a la CDD (Conserved Domains Database). Es tracta de llocs funcionals implicats en interacció amb ADN, ARN i proteïna. Beltran et al. s’interessen especialment per llocs funcionals que no es troben anotats en la CDD, i que són un total de 1942 llocs de 180 dominis: pensen que es tracta de llocs que fan de segona corona als primers en les interaccions corresponents.
Estabilitat proteica i patogenicitat
En la base de dades Human Domainome 1 hi ha 3652 variants amb anotacions clíniques conegudes. D’aquestes 621 són classificades com a patogèniques, 322 com a benignes i 2709 com a incertes. En total, hi ha 114 dominis amb alguna variant patogènica. Val a dir que el 75% de les 621 variants patogèniques es concentren en el 25% dels 114 dominis, i que n’hi ha 41 dominis amb una única variant patogènica.
En 380 de les 621 variants patogèniques (61%) Beltran et al. detecten desestabilització de domini. En 303 (48%) la desestabilització és forta. Aquestes proporcions són inferiors en les variants benignes (40% i 16%).
L’associació entre patogenicitat i desestabilització varia en diferents famílies de dominis. Per exemple, moltes mutacions patogèniques en cristal·lines β/γ que causen cataractes són fortament desestabilitzadores. En canvi poques ho són en homeodominis, dominis HMG-box o CUT, que són dominis d’unió a ADN.
En el domini LIM-2 de FHL1, la desestabilització és un bon classificador de variants patogèniques que provoquen miopatia corporal reductora, i que s’associen a l’acumulació muscular d’agregats de FHL1.
També tenen un caràcter desestabilitzador les variants patogèniques del domini SAM de TP63 implicades en la síndrome de fissura palatal, les quals s’associen a agregació de TP63.
En canvi, la majoria de mutacions del domini MBD del gen MECP2 que provoquen la síndrome de Rett dominant no són desestabilitzadores. En aquests casos n’hi ha prou amb un al·lel deficient perquè sigui insuficient la unió a ADN metilat de la proteïna MECP2.
Tampoc no semblen associades a l’estabilitat de proteïna les mutacions de l’homeodomini CRX que provoquen distròfies retinals.
Estabilitat de proteïnes i caràcter recessiu o dominant de mutacions patogèniques
La comparació de tots els dominis que presenten un mínim de 20 variants clíniques indica que l’estabilitat proteica explica bona part de la patogenicitat i de l’aptitud evolutiva.
En les malalties genètiques recessives la causa molecular sol ser una pèrdua de funció. En les malalties genètiques dominants hi solen haver mecanismes addicionals de guany o pèrdua de funció o efectes negatius d’agregació tòxica. Beltran et al. mostren que mentre en les malalties recessives l’estabilitat proteica explica un 44% de la variança, en les malalties dominant només ho fa en un 26%.
La conservació dels efectes mutacionals en proteïnes estructuralment homòlogues
L’assumpció de Beltran et al. és que una mutació en un domini proteic tindrà efectes anàlegs en totes les proteïnes que continguin aquest domini. No obstant, poden haver-hi variacions epistàtiques, és a dir relacionades en el context proteòmic i genòmic de la mutació.
Beltran et al. comproven aquesta darrera possibilitat calculant el canvi en l’energia de plegament que produeix cada mutació. En els 36 homeodominis estudiats els efectes mutacionals són força consistents.
En estendre l’anàlisi a totes les 26 famílies de dominis que contenen un mínim de 5 homòlegs en Human Domainome1, Beltran et al. troben una consistència semblant. Això indicaria que l’epistasi fa únicament una contribució petita a l’estabilitat proteica. L’aproximació dominiòmica seria efectiva per fer prediccions en proteïnes que comparteixin dominis.
Així doncs, Beltran et al. fan prediccions d’estabilitat per a 4.107.436 variants de 7.271 dominis. Això abasta 13.878 variants clíniques, de les quals 1310 són patogèniques, 951 benignes i 11617 de significat incert. Vora la meitat (52%) de les variants patogèniques reduirien l’estabilitat de proteïna, i un 34% ho farien fortament.
L’expansió de Human Domainome1 passaria, doncs, per mutagenitzar experimentalment exemples representatius de totes les famílies de dominis humans.
Lligams:
– Site-saturation mutagenesis of 500 human protein domains. Antoni Beltran, Xiang’er Jiang, Yue Shen, Ben Lehner. Nature (2025).
– El ‘Dominioma Humà’ explica la causa de diverses malalties hereditàries. CRG.eu, 08/01/2025.
