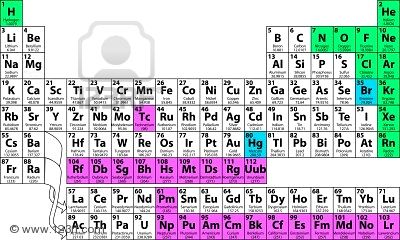
En certa manera, les quatre arrels o elements empedoclians sobreviuen en els nostres dies, de forma bastant directa, en els anomenats “quatre estats fonamentals d’agregació de la matèria”. De major a menor agregació, aquests estats són el sòlid (identificat amb la terra, amb partícules estretament vinculades, de manera que no poden moure’s lliurement, encara que sí vibrar), el líquid (identificat amb l’aigua, fluid incompressible, és a dir amb partícules de moviment lliure, però que retenen un volum constant amb independència de la pressió), el gas (identificat amb l’aire, fluid compressible, que tendeix a expandir-se pel moviment completament lliure de les partícules) i el plasma (identificat amb el foc, i que és un cas especial del gas, en el qual s’ha produït una dissociació entre els nuclis atòmics, de càrrega positiva, i els electrons, de càrrega negativa; és a dir, gas ionitzat). Com és habitual, però, la llista de fases de la matèria no és limita pas a quatre. Tot i amb tot, si ens fixem en les taules periòdiques dels elements, veurem que en moltes (un exemple) les caselles o els símbols químics reben colors d’acord amb la fase en la qual es troba cada element: el negre o el verd per als sòlids, el blau per als líquids i el vermell o el groc per als gasos. En d’altres, bo i mantenint, aquesta classificació s’afegeix un quart color per als elements “sintètics”, és a dir que no es troben en la natura (un exemple). No cal dir que la “fase” en aquestes classificacions es refereix a l’estat d’agregació de la matèria de la substància elemental en condicions estàndards de pressió i temperatura (definides habitualment com una temperatura de 298,15 K i una pressió de 105 Pa). Però hi ha d’altres classificacions o categoritzacions, com la basada en la distinció entre metalls i no-metalls. Els elements metàl•lics són aquells que presenten llustre, són bons conductors de calor i electricitat; són dúctils (i.e. se’n poden fer fils); són mal•leables (i.e. se’n poden fer làmines); són sòlids a temperatura ambient; i tenen la tendència a perdre un o més electrons. Val a dir que d’excepcions en trobem, car el mercuri és líquid a temperatura ambient, mentre el zinc no és ni mal•leable ni dúctil. Berzelius, que ja dubtava d’aquesta categorització, es limitava a diferenciar entre metalls i metal•loids (tot indicant, que els no-metalls també poden presentar propietats metàl•liques). No obstant, el mot metal•loid sol reservar-se per als elements amb propietats intermèdies (B, Si, Ge, As, Sb, Te, Po, At). En la taula periòdica dels elements, els metalls són majoria, mentre els no-metalls queden arraconats en el cantó superior dret, amb una franja separadora de metal•loids (exemple). La taula periòdica s’estructura en períodes i grups, i en principi cada grup d’element té unes característiques pròpies. De fet, la IUPAC té denominacions pròpies per al “grup 1” (els metalls alcalins), per al “grup 2” (els metalls alcalino-terris), per al “grup 15” (els pnictògens), per al “grup 16” (els calcògens), per al “grup 17” (els halògens) i per al “grup 18” (gasos nobles). Els altres grups, del 3 a l’11 (i de vegades amb el 12 inclòs), són anomenats per la IUPAC “metalls de transició”. De fet, els “metalls de transició” s’identifiquen amb el “bloc d” d’elements (de la mateixa manera, que el “bloc s” són els grups 1 i 2, i el “bloc p” són els grups 13-18). Fora dels “grups” reconeguts per la IUPAC trobem el “bloc f”, formar per lantànids (elements 57-71) i actínids (elements 89-103) que, juntament, amb l’escandi i l’itri, reben el nom col•lectiu de “elements de terres rares”. Una altra possible categorització d’elements faria: “metalls alcalins” (grup 1), “metalls alcalino-terris” (grup 2), “metalls de transició” (grups 3-12), “altres metalls” (els elements metàl•lics del bloc p), “metal•loids”, els “no-metalls poliatòmics” (que formen xarxes cristal•lines elementals, com fan el C, el P, el S i el Se), els “no-metalls diatòmics” (que, en estat estàndard, es presenten com a fluids diatòmics: H, N, O, F, Cl, Br, I), els “gasos nobles” (grup 18), els lantànids (període 6 del bloc f), els actínids (període 7 del bloc f) i els superactínids (bloc g). Encara n’hi ha més de classificacions, com la dissenyada per Victor Goldschmidt (1888-1947), que agrupa els elements químics segons la fase en la qual es troben preferentment a la Terra: litòfils (roques), sideròfils (associats al ferro), calcòfils (associats a minerals) o atmòfils (associats a l’atmosfera). Vés per on, de nou, quatre categories. En tot cas, nosaltres arribem a l’element 15 de la nostra sèrie.

Classificació tetrapartita dels elements d’acord amb la seva distribució en les diferents fases que els hostatgen en la Terra
Hennig Brand i la descoberta del fòsfor

Joseph Wright representava així el 1771 la descoberta del fòsfor, en el quadre titulat “L’alquimista a la recerca de la pedra filosofal”. El quadre exagera l’edat de Brand en descobrir el fòsfor i la lluïssor de l’element, però alhora retrata, des del punt de mira de la Il•lustració anglesa com la ciència moderna neix del pensament precientífic
Poc es coneix dels orígens d’Hennig Brand (de vegades escrit Brandt). Ell mateix després referiria l’alta posició de la seva família. Hom ha suposat que part dels seus coneixements pràctics els adquirí de ben jove com a aprenent de vidrier. Nascut devers el 1630, serví com a oficial en l’època anterior i directament posterior a la signatura del Tractat de Westfàlia (1648). Sembla que fou gràcies a la dot de la primera dona que, en retirar-se de l’exèrcit, encara relativament jove, va poder establir-se com a comerciant a Hamburg.
Però aviat la dedicació principal de Brand seria l’alquímia. La recerca de la pedra filosofal (lapis philosophorum) combinava el coneixement filosòfic amb la pràctica experimental i constituïa el Magnum Opus. La relació de la pedra filosofal amb la matèria primera, amb la crisopoeia (la capacitat de convertir els metalls innobles en metalls nobles) i amb la panacea (la substància per guarir tots els mals i assolir la perpetual joventut) era matèria d’innombrables publicacions, i no eren pocs els qui es llençaven, doncs, a la Magnum Opus, a la percaça de glòria, de riqueses, de la salvació terrenal i celestial.
La qüestió era trobar les combinacions adequades de materials. De moment, però, Brand no obtingué resultats. Vidu de la primera dona, el segon matrimoni, amb una vídua rica, Margaretha, li forní nous fons per continuar la recerca. Seguí els procediments descrits per l’estrasburguès F. T. Kessler a “400 Auserlensene Chemische Process”, que afirmava que era possible la conversió de metalls baixos a argent a través d’una barreja d’alum, salpetre i orina concentrada. Malgrat que trobà que la recepta no funcionava, decidí continuar els treballs amb orina.
Vers el 1669, en el decurs d’aquests treballs, Brand evaporava grans quantitats d’orina, i sotmetia a destil•lacions posteriors el residu sec obtingut, que quedava al fons de la retorta. Després escalfava la retorta fins que el residu es posava vermell. En una d’aquestes experiències, sobtadament, el residu començà a alliberar fums i flames, i desprendre un líquid. Traspassà el líquid resultà a un gerro i el tapà. En refredar-se se solidificà en un material blanc que fins i tot fred emetia una lluïssor d’un verd pàl•lid. Era a les fosques que millor es contemplava la lluïssor. El material alhora, en ser cremat, emetia una llum molt brillant.

Lluïssor del fòsfor blanc, visible en la foscor
Brand anomenà aquest material “phosphorus mirabilis” (admirable portador de llum). El mot “Phosphorus” prové, efectivament, de les arrels gregues, φως i φέρω, i havia estat emprat per l’Estel de l’Alba, és a dir Venus quan es troba en elongació oest. La forma llatina, Lucifer, també s’associava a aquest planeta, però a mitjan segle XVII ja es trobava fortament associada amb el Maligne.
Brand depurà el procés fins a poder descriure’n la recepta per fer “phosphorus mirabilis:
-1. Bullir orina (uns 5500 litres, en unitats modernes) fins que adopta la consistència d’un xarop dens.
-2. Escalfar aquest residu dens fins que destil•la un oli vermell, i separar aquest oli vermell.
-3. Refredar el residu, consisteix en una fase negra i esponjosa superior, i una part inferior constituïda per una sal blanca.
– 4. Descartar la sal, i barrejar l’oli vermell amb el material negre.
-5. Escalfar la barreja durant 16 hores, en el decurs de les quals es desprendran fums blancs, després un oli i finalment quedarà el fòsfor.
– 6. Deixar refredar el fòsfor fins que se solidifiqui en un material blanc (uns 120 grams, en unitats modernes).
Durant alguns anys, Brand conservà en secret aquesta descoberta. No la va començar a divulgar, primer entre gent de confiança, fins el 1675. El secret del Doctor Brand es va difondre aviat per Hamburg.
Johann Kunckel, més o menys de la mateixa edat de Brand, i que era apotecari de l’Elector de Saxònia, i que posseïa a Dresden un laboratori sota patrocini de l’Elector, s’interessà per la descoberta de Brand. Viatjà a propòsit des de Wittenberg a Hamburg. Brand li permeté veure el fòsfor. Kunckel feia algun temps que era interessat en la luminiscència de materials. La llum del fòsfor de Brand recordava a la de la pedra de Bolonya. Però mentre la pedra de Bolonya perdia la luminiscència si se la mantenia a les fosques per molt de temps, el fòsfor de Brand la retenia. Kunckel descrigué aquestes i altres propietats a Johann Daniel Kraft, un fabricant i comerciant de sedes de Leipzig amb notables coneixements alquímics, amb el qual havia fet amistat a Dresden. Kraft li guanyà la partida a Kunckel i, a través d’agents seus, va comprar la recepta a Brand per 200 tàlers.
Amb la recepta en el seu poder, Kraft va fer una gira per Europa presentant la substància. Parades obligades foren les corts reials, per començar la del duc Johann Friedrich de Saxònia. A aquesta demostració assistí, entre d’altres, Gottfried Leibniz, llavors de 30 anys. Leibniz, sabedor que la descoberta originària era de Brand, aprofità una visita a Hamburg el 1677 per entrevistar-s’hi. Brand, que tornava a tindre problemes econòmics per la insuficiència del seu negoci com a apotecari, vengué a Leibniz el secret de la confecció del fòsfor.
És molt possible que altres alquimistes, abans de Brand i tot, haguessin topat amb la substància. En tot cas, ara eren uns quants els qui anaven al darrera per reproduir-la. Kunckel, que havia trencat amb l’elector de Saxònia en el 1677, maldà per reproduir el mètode, i aconseguí de produir algunes quantitats (1678), encara que d’una qualitat inferior a la de Brand o Kraft.
Per la seva banda, Leibniz va fer en el 1678 una demostració del fòsfor i de la seva fabricació segons el mètode de Brand a Hannover. Leibniz recomanà al duc de Brünswick-Lüneburg la contractació de Brand per treballar en la Magnum Opus. El duc oferí a Brand sis mesos de salari per avançat. Encara que Brand també estudiava llavors una oferta del duc de Mecklenburg-Güstrow, es decidí finalment per Hannover. Després d’una estada en la que va fer fòsfor i en va fer demostracions, Brand tornà a Hamburg, però incomplí el compromís de fabricar més fòsfor. En el 1679, Brand tornà a Hannover, i arran d’aquesta estada Leibniz va publicar un tractar sobre la fabricació de fòsfor.
Un altre alquimista cèlebre que havia mostrat interès pel fòsfor, des del 1677, era Robert Boyle. Boyle tenia llavors 50 anys, i era un exemple del que podríem anomenar “quimista escèptic” (per fer servir el títol d’una de les seves obres, del 1661, The Sceptical Chymist: or Chymico-Physical Doubts & Paradoxes) i sostenia una visió atomista de la matèria, segons les qual els fenòmens naturals s’explicarien per la col•lisió de partícules en moviment. Boyle, en correspondència amb Kraft, no havia pogut arrencar-li més que la confessió que el fòsfor era fabricat a partir de “quelcom que pertany al cos humà”. Boyle hi va fer diversos intents a partir de femta i d’orina humanes, per ell mateix i contractant després els serveis de diversos alquimistes alemanys que residien a Londres, entre ells, Johann Joachim Becher (1635-1682). Fou finalment un jove alquimista, de 19 anys, natural de Nienburg, Saxònia, anomenat Ambrose Gottfried Hanckwitz, qui després de fer una visita a Hamburg a Brand, aconseguí per a Becher i Boyle el secret del procés. El jove, que a Anglaterra adoptaria el nom d’Ambrose Godfrey, aconseguí produir fòsfor a Londres en el 1680. Boyle millorà el procés en afegir sorra a l’orina. Alhora, en el mateix any, Boyle mostrà com el fòsfor es podria emprar per encendre escuradents als qual s’hagués aplicat sofre en la punta. Boyle atribuí la lluïssor del fòsfor a la debilitació de l’aire amb el qual entra en contacte.
En el 1682, Godfrey començà, amb l’ajut financer de Boyle, un projecte per fabricar fòsfor a escala comercial. En el 1685, ja disposava d’un forn domèstic i emprava com a matèria primera orina i femtes humanes de la veïna Bedford House. Venia fòsfor blanc (a 50 xílings a l’engròs o 60 xílings al detall) però també fòsfor líquid (obtingut de barrejar fòsfor amb oli d’orina). De totes formes, el negoci del fòsfor no li donava per viure, i els seus ingressos principals provenien de la seva feina de director de laboratori a l’Apothecaries Hall.
Brand es va morir el 1692. En aquella època, la producció de fòsfor ja era dominada en el pla comercial per Godfrey. Godfrey sempre maldà per mantindre en secret tan allò que havia après de Brand, Becher i Boyle, com les millores que havia introduït en el procediment. A començament del segle XVIII, el laboratori de Godfrey produïa 50 lliures anuals, amb una facturació de 2000 lliures esterlines. En el 1707, Godfrey llogà un nou establiment al mateix carrer Southampton, on obrí una farmàcia i s’instal•là ell i la seva família. El fill gran, anomenat Boyle en honor de l’antic mestre, fou alquimista dedicat, però sense sentit per al negoci. Així, quan Ambrose Godfrey es va morir a 81 anys el 15 de gener del 1741, van haver de ser els seus dos germans menors, Ambrose i John, els qui es feren a càrrec del negoci a partir del 1742, mentre es comprometien a passar a una pensió al germà gran. El negoci familiar ja es basava fonamentalment en el fòsfor i, malgrat que tenia clients a tot Anglaterra i al continent, va fer fallida en el 1746. El fill de Boyle Godfrey, anomenat Ambrose com l’avi, se’n va fer càrrec, amb més èxit que no pas la generació anterior.
En el 1769, Johan Gottlieb Gahn (1745-1818) i Carl Wilhelm Scheele (1742-1786) descrigueren químicament la fracció mineral dels ossos com una sal fosfòrica i calcària. En els anys següents, es feren diversos intents per produir fòsfor a partir dels ossos, i fou Bertrand Pelletier (1761-1797) qui reeixí finalment a fer-ho, uns 10 anys després de la descoberta de Gahn i Scheele. El mètode de Pelletier consistia en rostir els ossos, i hom abocava el contingut a retortes d’argila que es col•locaven a un forn a alta temperatura. En una altra variant, hom produïa fosfats a partir d’ossos submergits en àcid.

Antoine Lavoisier
Antoine Lavoisier reconegué el caràcter de substància elemental o simple del fòsfor ja en el 1777. En els seus treballs sobre la combustió, la combustió del fòsfor és especialment esmentada. Així, en un article publicat a les Mémoires de l’Académie Royale des Sciences (592-600, 1777) llegim:
“En tota combustió el cos cremat canvia a àcid amb l’addició de la substància que augmenta el seu pes. (…). Si es crema fòsfor, el producte de la combustió és àcid fosfòric”.
Lavoisier contrasta la calcinació dels metalls, lenta, amb la calcinació del sofre i del fòsfor, “gairebé instantània”. Això facilita que s’evidenciï el fet que, en la combustió, “el fòsfor absorbeix aire o, més aviat, la base de l’aire, de la qual cosa en resulta l’àcid fosfòric”. L’explicació de Lavoisier contradeia la idea de la teoria del flogist, és a dir que hi hagués una immensa quantitat de foc fix, anomenat combustible, en les matèries oxidables o inflamables, inclosa el fòsfor, la idea que precisament havia sostingut en el seu moment Becher.
Lavoisier també s’ocupà de la lluminiscència del fòsfor, i comprovà com aquesta lluminiscència es perdia si el fòsfor era en contacte amb oxigen pur per comptes d’aire.
El fòsfor apareix, doncs, en la llista de 33 substàncies simples del “Tractat elemental de química” (1789) de Lavoisier. El fòsfor és comptat en la categoria de “substàncies simples no-metàl•liques oxidables o acidificables”, en la qual també trobem el sofre, el carboni, el radical muriàtic, el radical fluòric i el radical boràcic. Lavoisier explicar l’àcid fosfòric com una substància composta de fòsfor, hidrogen i oxigen, mentre que la base de l’àcid fosfòric, el fosfat, seria una combinació de fòsfor i oxigen.
Ja hem esmentat que Boyle havia defensat ja una teoria atòmica de la matèria en el 1661. La sistematització de la teoria atòmica, però, no acabaria d’arrencar fins que l’enuncià John Dalton en el 1803. En el “New System of Chemical Philosophy”, Dalton presenta el fòsfor com un element simple, i el simbolitza amb un estel de tres puntes.
En el 1814, Jöns Jacob Berzelius fa una llista de 47 substàncies elementals. Com que el fòsfor és un metal•loid (i.e., un no-metall), Berzelius el simbolitza senzillament amb la inicial de la forma llatina, Phosphorus, P.
El desplegament de la química elemental, d’altra banda, posava de manifest la importància del fòsfor, en forma de fosfats, en la fertilitat dels sòls. Juntament amb el nitrogen i el potassi, el fòsfor apareixia com un dels factors limitants en el creixement vegetal. Ja en el 1828, Carl Sprengel formulava la “llei del mínim”, que popularitzaria després Justus von Liebig (1803-1873), segons la qual la taxa de creixement vegetal depèn únicament d’aquell factor nutricional que es trobi en menor abundància. El fòsfor, en molts sòls, era aquest factor nutricional. Liebig deplorava la separació creixent de la població humana respecte del camp, ja que tallava la reintroducció de nitrogen i fòsfor als sòls a través de la femta i de l’orina.
D’altra banda, ara que el fòsfor no es trobava ja sota secret comercial de cap mena, creixien a Anglaterra i altres països una indústria de producció de fòsfor. El fòsfor produït era alhora matèria primera per a les indústries dels llumins, que encarnaven aquella observació feta per Boyle en el 1680. Els treballadors d’aquestes indústries, incloent-hi infants, eren exposats a la toxicitat del fòsfor. Els primers llumins de fòsfor es van produir a partir dels anys 1830, i en la formulació contenien una barreja de fòsfor blanc, addicionada amb adhesiu i un donador d’oxigen (KCl, PbO2, nitrats, etc.).
En el anys 1840, el guano esdevenia una font de fosfats per a l’agricultura (fertilitzants) i per a la indústria i, alhora, en una font de fòsfor, que acabà per desplaçar els ossos i la cendra d’ossos. En els anys 1850, a més, s’aconseguí l’obtenció comercial de fòsfor a partir de roques fetes de fosfats (particularment, de fosfat càlcic).
D’acord amb la llei de l’octet, formulada per John Newlands, en el 1865, el fòsfor formava part, amb nombre atòmic de 13, del grup d’elements encapçalat pel nitrogen. La cosa tenia sentit si atenem a algunes propietats bàsiques de tots dos elements i era enfortida pel rol de tots dos com a nutrients limitants habituals del sòl. De manera independent, la taula periòdica de Dmitri Mendeleev, del 1869, fa la mateixa relació. El fòsfor, en la versió d’aquesta taula del 1871, apareix en el període 3 del grup V d’elements, caracteritzat per formar trihidrurs (RH3) i pentaòxids divalents (R2O5). En les formes ja més definitives de la taula periòdica, com la de Henry Moseley, del 1913, s’hi introduirien canvis, però el fòsfor ja no es mouria de lloc.

La confecció de llumins o mistos, en emprar fòsfor blanc i d’altres substàncies, generava una exposició crònica continuada entre les treballadors (majoritàriament, noies i dones joves). A això s’afegien salaris de misèria (deduïts, a més, a base de multes), llargues jornades de treball i l’amenaça continuada de perdre la feina
La producció de fòsfor i llumins continuà en augment. La toxicitat del fòsfor era ben coneguda, i casos d’intoxicacions agudes mortals, per accident, per suïcidi o, fins i tot, per assassinat, eren reportades. Dels efectes de l’exposició crònica n’eren conscients els treballadors de les indústries que produïen fòsfor o en treballaven. A Anglaterra, hom encunyà l’expressió “phossy jaw” per referir-se a una malaltia professional lligada a l’exposició a vapors de fòsfor, i que es manifestava en mals de queixal i en gingivitis, deguts a una necrosi mandibular. El lligam era manifest en les autòpsies, quan els ossos afectats manifestaven una lluïssor de color verdosa a la foscor. Aquesta intoxicació crònica i els seus efectes foren un element detonant en vagues, com la que protagonitzaren les treballadores de Bryant and May Factory (Bow, Londres) el juliol del 1888. A resultes d’aquesta vaga, Annie Besant (1847-1933) esdevingué una de les impulsores de la campanya per prohibir l’ús del fòsfor blanc.
La prohibició d’emprar fòsfor blanc en els llumins ja havia començat en algunes jurisdiccions en el 1872. El 1906, se signà la “Convenció Internacional respecte la prohibició de l’ús del fòsfor blanc (groc) en la manufactura de llumins”, coneguda com la Convenció de Berna. El procés de prohibició, però, continuà fins el 1925. Com a substituts del fòsfor blanc, s’empra des de llavors sesquisulfur de sofre (P4S3), sofre (S) o sulfur d’antimoni. Mentre els llumins de fòsfor blanc es podien encendre fregant-los en qualsevol superfície, els nous “llumins de seguretat” requereixen normalment una superfície rugosa rica en fòsfor vermell, introduïda en la mateixa capsa de llumins.
La introducció del forn d’arc elèctric submergit, sobretot a partir dels anys 1890, van fer més atractives les roques riques en fosfats com a font de fosfats i fòsfor, que desplaçaren definitivament tant les cendres d’ossos com el guano (les reserves del qual començaven a exhaurir-se). L’augment consegüent de la producció de fòsfor va fer que s’interessés la indústria militar. Durant la Primera Guerra Mundial (1914-1918) s’emprà fòsfor blanc en artefactes incendiaris, i també per generar cortines de fum i traçar la trajectòria de projectils. Els horrors de les bombes incendiàries de fòsfor es repetirien, amb escreix, a la Segona Guerra Mundial.
Vam veure en el seu moment, com George Gabriel Stokes, en el 1852, encunyà la paraula fluorescència, a partir de la fluorita, per designar l’emissió de llum visible per part d’una substància que rep radiació ultraviolada. Per analogia, hom creà el mot fosforescència per referir-se a la luminiscència del fòsfor. A diferència dels materials fluorescents, els materials fosforescents no reemeten immediatament en forma de radiació l’energia rebuda, sinó que ho fan gradualment en el temps.
Fluorescència i fosforescència foren explicades amb el desplegament de la mecànica quàntica. Són dos tipus de fotoluminescència, iniciada per la fotoexcitació (excitació per absorció de radiació electromagnètica) del material, i que, a través, de processos diferents de relaxació, comportà una reemissió de l’energia absorbida.
Però la luminiscència del fòsfor blanc no encaixava bé en aquest esquema. El fòsfor blanc només presentava luminiscència en certes condicions de temperatura, pressió i composició de l’aire amb el qual es troba en contacte. Diverses recerques, assenyalaren que el fòsfor blanc no era pas un material fosforescent, sinó quimioluminiscent, és a dir que la luminiscència era el resultat de la reacció química del vapor de fòsfor amb oxigen. Una explicació satisfactòria no arribà fins el 1974, quan Richard J. VanZee i Ahsan U. Khan oferí el mecanisme: el fòsfor sòlid o líquid, en contacte amb una atmosfera adequada d’oxigen, reacciona formant-hi HPO i P2O2, substàncies efímeres responsables en darrer terme de la luminiscència del fòsfor. Tot i amb tot, el mot fosforescència ha quedat.

Un exemple de substància veritablement fosforescent és la pols d’òxid de silicat aluminat d’estronci i europi
El fòsfor: isòtops i abundància
La massa atòmica estàndard del fòsfor, de 30,973761998 uma, és la del seu únic isòtop estable, el 31P. Podem, però, fer un llistat de tots els isòtops que han estat detectats i estudiats:
– fòsfor-24 (24P; 24,03435 uma). Nucli format per 15 protons i 9 neutrons. És un isòtop molt inestable. Decau normalment (>99,9%) a silici-23 (amb emissió d’un protó) o, alternativament (<0,01%), a silici-24 (amb emissió d’un positró).
– fòsfor-25 (25P; 25,02026 uma). Nucli format per 15 protons i 10 neutrons. És un isòtop molt inestable, amb una semivida de menys de 3•10-8 s. Decau a silici-24, amb emissió d’un protó.
– fòsfor-26 (26P; 26,01178 uma). Nucli format per 15 protons (un d’ells en halo) i 11 neutrons. És un isòtop molt inestable, amb una semivida de 0,0437 s. Decau normalment (98,1%) a silici-26 (amb emissió d’un positró) o, alternativament, a magnesi-24 (1,0%; amb emissió de dos protons i d’un positró) o a alumini-25 (0,9%; amb emissió d’un protó i d’un positró).
– fòsfor-27 (27P; 26,999230 uma). Nucli format per 15 protons i 12 neutrons. És un isòtop molt inestable, amb una semivida de 0,260 s. Decau normalment (99,93%) a silici-27 (amb emissió d’un positró) o, alternativament (0,07%) a alumini-26 (amb emissió d’un protó i d’un positró).
– fòsfor-28 (28P; 27,992315 uma). Nucli format per 15 protons i 13 neutrons. És un isòtop molt inestable, amb una semivida de 0,2703 s. Decau normalment (99,99%) a silici-28 (amb emissió d’un positró) o, alternativament, a alumini-27 (0,0013%; amb emissió d’un protó i d’un positró) o a magnesi-24 (0,00086%; amb emissió d’un nucli de 4He i d’un positró).
– fòsfor-29 (29P; 28,9818006 uma). Nucli format per 15 protons i 14 neutrons. És un isòtop inestable, amb una semivida de 4,142 s. Decau, amb emissió d’un positró, a silici-29.
– fòsfor-30 (30P; 29,9783138 uma). Nucli format per 15 protons i 15 neutrons. És un isòtop inestable, amb una semivida de 149,9 s (2 minuts i mig). Decau, amb emissió d’un positró, a silici-30.
– fòsfor-31 (31P; 30,97376163 uma). Nucli format per 15 protons i 16 neutrons. És l’únic isòtop estable de fòsfor. La pràctica totalitat dels àtoms de fòsfor són de 31P.
– fòsfor-32 (32P; 31,97390727 uma). Nucli format per 15 protons i 17 neutrons. És un isòtop inestable, amb una semivida de 1,2323•106 s (14 dies i 6 hores). Decau, amb emissió d’un electró, a sofre-32. Hi ha una petita taxa de producció natural que fa que sigui present en traces en mostres ambientals. Aquest radioisòtop pot ésser generat també sintèticament a través del bombardeig amb neutrons de 32S (32S + n -> 32P + 1H). Se l’ha emprat en estudis sobre metabolisme del fòsfor en els éssers vius i, molt particularment, en radiomarcatge d’àcids nucleics, en experiments cabdals per al desenvolupament de la biologia molecular en els anys 1950 i 1960. El 32P també és emprat en medicina nuclear, per exemple com a traçador de tumors malignes, que tenen una tendència a acumular més fosfat que no pas els teixits normals). Aquesta mateixa propietat ha fet que se l’hagi investigat com a component en tractament quimioteràpics (Pattillo et al., 1995). Com que és un radioisòtop beta-emissor d’alta energia (1,71 MeV), cal extremar les mesures de seguretat per evitar l’exposició a les radiacions i al mateixradioisòtop.
– fòsfor-33 (33P; 32,9717255 uma). Nucli format per 15 protons i 18 neutrons. És un isòtop inestable, amb una semivida de 2,189•106 s (uns 26 dies). Decau, amb emissió d’un electró, a sofre-33. Com que és un radioisòtop beta-emissor menys energètic que el 32P (0,25 MeV), hom l’ha emprat en el radiomarcatge de nucleòtids en aplicacions com la seqüenciació d’ADN o en tècniques que requereixen una millor resolució de marcatge.
– fòsfor-34 (34P; 33,973636 uma). Nucli format per 15 protons i 19 neutrons. És un isòtop inestable, amb una semivida de 12,43 s. Decau, amb emissió d’un electró, a sofre-34.
– fòsfor-35 (35P; 34,9733141 uma). Nucli format per 15 protons i 20 neutrons. És un isòtop inestable, amb una semivida de 47,3 s. Decau, amb emissió d’un electró, a sofre-35.
– fòsfor-36 (36P; 35,978260 uma). Nucli format per 15 protons i 21 neutrons. És un isòtop inestable, amb una semivida de 5,6 s. Decau, amb emissió d’un electró, a sofre-36.
– fòsfor-37 (37P; 36,97961 uma). Nucli format per 15 protons i 22 neutrons. És un isòtop inestable, amb una semivida de 2,31 s. Decau, amb emissió d’un electró, a sofre-37.
– fòsfor-38 (38P; 37,98416 uma). Nucli format per 15 protons i 23 neutrons. És un isòtop molt inestable, amb una semivida de 0,64 s. Decau majoritàriament (88%) a sofre-38 (amb emissió d’un electró) o, alternativament (12%) a sofre-37 (amb emissió d’un neutró i d’un electró).
– fòsfor-39 (39P; 38,98618 uma). Nucli format per 15 protons i 24 neutrons. És un isòtop molt inestable, amb una semivida de 0,190 s. Decau majoritàriament (74%) a sofre-39 (amb emissió d’un electró) o, alternativament (26%) a sofre-38 (amb emissió d’un neutró i d’un electró).
– fòsfor-40 (40P; 39,99130 uma). Nucli format per 15 protons i 25 neutrons. És un isòtop molt inestable, amb una semivida de 0,153 s. Decau majoritàriament (70%) a sofre-40 (amb emissió d’un electró) o, alternativament (30%) a sofre-39 (amb emissió d’un neutró i d’un electró).
– fòsfor-41 (41P; 40,99434 uma). Nucli format per 15 protons i 26 neutrons. És un isòtop molt inestable, amb una semivida de 0,100 s. Decau majoritàriament (70%) a sofre-41 (amb emissió d’un electró) o, alternativament (30%) a sofre-40 (amb emissió d’un neutró i d’un electró).
– fòsfor-42 (42P; 42,00101 uma). Nucli format per 15 protons i 27 neutrons. És un isòtop molt inestable, amb una semivida de 0,0485 s. Decau bé a sofre-42 (50%, amb emissió d’un electró) o a sofre-41 (50%, amb emissió d’un neutró i d’un electró).
– fòsfor-43 (43P; 43,00619 uma). Nucli format per 15 protons i 28 neutrons. És un isòtop molt inestable, amb una semivida de 0,0365 s. Decau a sofre-42, amb emissió d’un neutró i d’un electró.
– fòsfor-44 (44P; 44,01299 uma). Nucli format per 15 protons i 29 neutrons. És un isòtop molt inestable, amb una semivida de 0,0185 s. Decau a sofre-44, amb emissió d’un electró.
– fòsfor-45 (45P; 45,01922 uma). Nucli format per 15 protons i 30 neutrons. És un isòtop molt inestable, amb una semivida de l’ordre de 0,001 s. Decau a fòsfor-45, amb emissió d’un electró.
– fòsfor-46 (46P; 46,02738 uma). Nucli format per 15 protons i 31 neutrons. És un isòtop molt inestable, amb una semivida de l’ordre de 0,001 s. Decau a fòsfor-46, amb emissió d’un electró.
L’àtom neutre de fòsfor conté 15 electrons, amb una configuració basal d’escorça de 1s22s22p63s23p3. És, doncs, l’element del tercer període per al grup 15 (pnictògens, = grup VA o VB, en altres terminologies). Dins del grup, és considerat un no-metall, com el nitrogen, però mentre el nitrogen és classificat entre els “no-metalls diatòmics”, el fòsfor entra en el grup dels “no-metalls poliatòmics”. El nombre d’oxidació més habitual és el +5, per bé que, de fet, forma compostos també amb +4, +3, +2, +1 (p.ex. PI), -1, -2 i -3. Presenta un radi de van der Waals de 1,8•10-10 m.
El fòsfor en estat elemental es presenta com un sòlid en condicions estàndards. Se n’han descrit diversos al•lòtrops sòlids:
– el fòsfor blanc, també conegut com a fòsfor groc i tetrafòsfor (P4), és l’al•lòtrop descobert originàriament per Hennig Brand en el 1669. El nom de fòsfor groc fa referència a la tonalitat que adquireix quan és exposat a la llum. Ja hem vist com, en la foscor, si és exposat a un determinat rang de pressió parcial de gas oxigen, emet una lluïssor verdosa. Per la seva toxicitat i inflamabilitat, sol presentar-se en forma de pastilles recobertes de pentòxid de difòsfor (P4O10) conservades immergides en aigua. Cristal•logràficament, hom diferencia:
+ forma α (fòsfor blanc-α). És la forma estable en condicions estàndards de pressió temperatura. Consisteix en una xarxa cristal•lina cúbica centrada en el cos.
+ forma β (fòsfor blanc-β). Consisteix en una xarxa cristal•lina hexagonal. En condicions estàndards de pressió, la forma α esdevé reversiblement forma β en baixar de 195,2 K.
– el fòsfor vermell. El fòsfor blanc, en escalfar-se en condicions estàndards de pressió a temperatures de més de 520 K, esdevé fòsfor vermell. També l’exposició del fòsfor blanc a la radiació solar pot tornar-lo fòsfor vermell. Consisteix en una xarxa amorfa d’àtoms de fòsfor, per bé que a més altes temperatures cristal•litza. Una vegada obtingut el fòsfor vermell a altes temperatures, es manté aquest al•lòtrop, que és molt menys inflamable que el fòsfor blanc a temperatures inferiors a 500 K.
– el fòsfor violeta. Fou descrit originàriament el 1865 per Johann Wilhelm Hittorf (1824-1914), quan escalfà fòsfor vermell en un tub segellat a una temperatura de 800 K. Consisteix en cristalls monoclínics o romboèdrics opacs i brillants (d’ací el nom de fòsfor metàl•lic).
– el fòsfor negre. S’obté del fòsfor blanc escalfant-lo a pressions elevades (1,2 GPa). En condicions estàndards de pressió i temperatura, és l’al•lòtrop de fòsfor elemental termodinàmicament més estable. Recorda pel color i la textura al grafit. Consisteix en una xarxa cristal•lina ortoròmbica. Com en el cas del grafè i del silicè, és possible preparar fosforè, una monocapa cristal•lina de fòsfor, d’interès com a semiconductor (Liu et al., 2014)

Els quatre al•lòtrops més comuns del fòsfor (a dalt) són el fòsfor blanc, el fòsfor vermell, el fòsfor violeta i el fòsfor negre. Les relacions esquemàtiques, segons pressió i temperatura, dels al•lòtrops es representen a sota

El punt de fusió depèn de l’al•lòtrop. Mentre el fòsfor blanc es fon a 317,4 K a pressió estàndard, el fòsfor negre no ho fa fins a 880 K. El fòsfor vermell i el fòsfor violeta no es fonen sinó que sublimen a gas, respectivament, a 689 K i 890 K, a pressió estàndar. El punt d’ebullició del fòsfor blanc, a pressió estàndard, és de 554 K. En estat gasós, el fòsfor elemental pot trobar-se de manera monoatòmica o diatòmica (P2). En condicions estàndards de pressió, el fòsfor diatòmic és forma termodinàmicament estable del fòsfor elemental a temperatures de 1500-2300 K.
Com correspon als elements químics amb un nombre de protons imparell, el fòsfor és menys abundant que els dos elements que el precedeixen (14Si) i segueixen (16S) en la taula periòdica. L’abundància depèn de la taxa neta amb la qual el 31P és produït en estels massius. Dels elements més lleugers, el fòsfor únicament supera en abundància el fluor, el liti, el bor i el beril•li. En canvi, la llista d’elements més pesants que el superen en abundància és llarga: a banda del sofre, l’argó, el calci, el crom, el manganès, el ferro i el níquel.
El fòsfor pot adoptar múltiples nombres d’oxidació, la qual cosa comporta un ventall considerable de compostos. L’ió fosfur (P3-) pot combinar-se amb l’hidrogen per donar lloc a la fosfina (PH3); en meteorits el trobem combinat en forma de schreibersita [(Fe,Ni)3P]. Un altre compost amb l’hidrogen és la difosfina (P2H4).
Dels oxoàcids del fòsfor, els més freqüents són l’àcid hipofosforós (H3PO2), l’àcid ortofosforós (H3PO3) i l’àcid ortofosfòric (H3PO4), en els quals, respectivament, el fòsfor es troba amb un estat d’oxidació de +1, +3 i +5. Els compostos més freqüents del fòsfor són derivats de l’ió fosfat, PO4sup>3-. Els compostos organofosforats es basen en l’enllaç P-C o, més freqüentment, en els enllaços P-O-C.
En la Terra, l’abundància atòmica del fòsfor és del 0,102% (superada, doncs, per l’oxigen, el magnesi, el silici, el ferro, l’alumini, el calci, el níquel, l’hidrogen, el sofre, el crom, el sodi i el carboni). En termes de massa, el percentatge és de 0,121%. El fòsfor és un element formador de roques (litòfil en la classificació de Goldschmidt), per bé que amb una rellevància secundària respecte dels elements més abundants de litosfera (oxigen, silici, alumini, sodi, calci, potassi i ferro). En termes de massa, l’abundància en l’escorça és del 0,12%, similar al valor que trobem en el mantell i en el nucli.
Dins de l’escorça terrestre, el fòsfor es troba majoritàriament en forma de fosfats minerals, com ara les apatites: hidroxilapatita (Ca10(PO4)6(OH)2), fluorapatita (Ca10(PO4)6F2) i clorapatita (Ca10(PO4)6Cl2).
La solubilitat dels ions fosfat fa que el fòsfor sigui present en la hidrosfera, si bé amb una abundància molt menor que en la litosfera. En els oceans, la concentració és de 60-90 mg•m-3. En l’atmosfera es troba tan sols en forma de traça.
En la biosfera, en canvi, la concentració de fòsfor és un ordre de magnitud superior a la de la litosfera. El fòsfor és un dels macroelements biològics. El trobem en forma de fosfats, per una banda dissolts en els fluids biològics (fosfats inorgànics, Pi) i per l’altra units covalentment a biomolècules. El fòsfor és element obligat en els àcids nucleics, i de fet els nucleòtids que formen les cadenes nucleiques es troben unides per enllaços fosfat.

Estructura molecular del monofosfat de desoxiadenosina (dAMP), un nucleòtid.
El fòsfor també és element obligat dels fosfolípids, constituents bàsics de les membranes cel•lulars.

Esquema de la bicapa de fosfolípids que és la base estructural de les membranes biològiques. El fòsfor ocupa un lloc central en l’estructura molecular del fosfolípid, unint la part de la molècula amb afinitat amb aigua amb la part hidròfoba
L’enllaç que uneix el grup fosfat amb les molècules orgàniques té una elevada energia. Hom coneix amb el nom genèric de fosfàgens, les molècules orgàniques que presenten grups fosfats. El trifosfat d’adenosina (ATP) fou identificat en el 1941 per Fritz Albert Lipmann (1889-1986) com la molècula intermediària entre les reaccions exergòniques de la cèl•lula i les reaccions endergòniques. Així, el metabolisme degradador (catabolisme) produeix energia que és vehiculada, si més no en part, per la reacció ADP + PO43+ -> ATP. El metabolisme sintètic (anabolisme) empra la reacció ATP -> ADP + PO43+ per afavorir reaccions que requereixen una entrada d’energia.
El grup fosfat també és un grup prostètic de les proteïnes, concretament a través dels residus de tirosina, serina i treonina. La fosforilació i defosforilació de proteïnes en llocs específics de la molècula constitueix un mecanisme reversible de modulació de la seva activitat, i té una importància considerable en les vies de senyalització intracel•lular.
A banda, els fosfats participen en l’estructura d’importants minerals biogènics. El component principal dels ossos, per exemple, és la hidroxiapatita, un fosfat càlcic. Una part substancial dels minerals fosfats de l’escorça tenen un origen fòssil, és a dir que són el resultat de la transformació de biominerals.
En el cos humà, el fòsfor suposa, en termes de massa, un 1,2% (únicament separat pels quatre grans bioelements, O, C, H i N; i pel Ca). En termes atòmics, suposa un 0,22%. En el cos humà, la majoria del fòsfor (85-90%) es troba com a constituent de l’apatita dels ossos i de l’esmalt dentari. La concentració en sang és d’un 0,4 kg•m3+, del qual un 70% es troba en forma de fòsfor orgànic, i un 30% d’inorgànic (cations fosfat i pirofosfat). S’adquireix en la dieta, fonamentalment a través de fosfoproteïnes (làctics, càrnics, ous, cereals, llegums, fruits secs), amb una taxa de consum i excreció diària de 1-3 grams. L’excreció es realitza en forma de cations fosfat (H2PO4–, HPO42-), a través de l’orina i de la femta. La deficiència nutricional en fòsfor pot ésser conseqüència d’una ingesta deficient (en dietes pobres en proteïna i calci), de problemes en l’absorció intestinal de fosfats o d’un excés d’excreció urinària. En resulta una hipofosfatèmia, que es manifesta inicialment en disfuncions musculars i neurològiques. La hiperfosfatèmia, d’altra banda, pot conduir a una calcificació de teixits i pot interferir amb el metabolisme del ferro, calci, magnesi i zinc.
Per a tots els organismes, doncs, el fòsfor és un macromineral essencial. Els fosfats poden ésser el factor limitant de molts ecosistemes, la qual cosa implica que un augment pot comportar problemes per eutrofització (sobrecreixement algal en ecosistemes aquàtics, etc.).
El cicle del fòsfor
El cicle biogeoquímic del fòsfor presenta tota una sèrie de particularitats. En primer lloc, es diferencia d’altres bioelements, pel fet que majoritàriament en la biosfera el trobem en un estat d’oxidació. Les reaccions d’oxo-reducció que tan rellevants són en el cicle del carboni (del CH4 al CO2), del nitrogen (del NH4+ al NO3–, passant pel N2), de l’oxigen (de l’H2O al O2) o del sofre (de l’H2S al SO42-), no apareixen de manera rellevant en el cas del fòsfor, que es troba tothora en forma fosfat (PO43-), integrat en minerals (apatita), unit covalentment a molècules orgàniques o dissolt en la fase aquosa. En segon lloc, en el cicle del fòsfor, el compartiment atmosfèric és pràcticament menystenible. Finalment, el cicle del fòsfor és obert en molts ecosistemes, i hi ha una tendència secular a la pèrdua de fòsfor disponible per als ecosistemes continentals; tendència que tan sols es contrapesada per processos geològics de llarguíssima durada.

El cicle del fòsfor en un ecosistema aquàtic. Per escolament, hi ha una aportació constant de fosfat des del medi emergit al medi aquàtic (actualment s’estima l’escolament de fòsfor cap als ecosistemes marins entre 8-15 milions de tones). Aquesta aportació exògena de fòsfor inorgànica, augmentada per l’activitat humana, és incorporada pels productors primaris en forma de fòsfor orgànic, que és aprofitat pels predadors. El fòsfor orgànic és reciclat pels organismes descomponedors. Els moviment de la columna d’aigua permeten la recuperació del fòsfor, si bé una part pot acabar segrestada en els sediments. Aquesta darrera part únicament serà mobilitzada per transformacions geològiques.
El recanvi del fòsfor que trobem en els organismes és ràpid, particularment en aquells que no el retenen per a constituir biominerals. En contrast amb aquesta rapidesa, els moviments en la hidrosfera, particularment en els oceans, o, encara més, en la litosfera, són molt més lents.
Els fosfats en forma de roques, sobretot apatita, no són utilitzables per a la majoria d’organismes. Cal un procés de meteorització, biològica o abiòtica, per tal que sigui utilitzable pels productors primaris (plantes, algues). En el sòl, els fosfats es “fixen” o “immobilitzen” en ser absorbits per òxids de ferro, hidròxids d’alumini, superfícies argiloses o partícules de matèria orgànica. D’aquest reservori immobilitzat, plantes i fongs el poden solubilitzar i absorbir; alhora la descomposició de matèria orgànica morta comporta un reciclatge de fosfats. L’erosió de sòls comporta una pèrdua constant de fosfats cap als sistemes aquàtics, els quals també cedeixen fosfats a través de la matèria orgànica sedimentada. Són processos geològics els que convertiran el fosfat sedimentat en fosfat de roca, i els que tornaran a exposar el fosfat de roca a la meteorització, tancant a la llarga el cicle.
Justus von Liebig (1803-1873) ha estat considerat el “pare de la indústria dels fertilitzants”. Von Liebig popularitzà el concepte de “nutrient limitant”, i posà de manifest com el rendiment de sòls i cultius en l’agricultura era degut a una insuficiència elemental. El fòsfor, el nitrogen o el potassi eren els “nutrients limitants més comuns”. Alhora, Liebig alertava de les conseqüències que tenia per a la fertilitat dels sòls el procés de concentració de la població humana en ciutats. La separació metabòlica entre els llocs de producció d’aliments (camp) i els llocs de consum (ciutat) provoca efectivament un traspàs de materials i, en conseqüència, també, un traspàs de fòsfor. Si en les societats preindustrials, els éssers humans retornaven al medi local, a través de femtes i orines, el fòsfor consumit, ara la concentració humana significava un abocament de fosfats en punts concrets, la qual cosa facilita la seva pèrdua concentrada cap a ecosistemes marins. Els abocaments d’aigües residuals, rics en fosfats, produeixen alteracions eutrofitzants en els punts de destí. Però, tal com alertava, Von Liebig la retirada de fosfats amenaçava la fertilitat dels sòls.
La qüestió, és clar, era més complexa. Revolució industrial i revolució agrícola són ben entrelligades en les transformacions socials que descrivia Von Liebig. Les innovacions agrícoles feien sobrera una mà d’obra que era disponible per a la gran indústria. L’expansió dels mercats, a Europa i a Ultramar, estimulà la producció agrícola. La qüestió de la millora dels sòls fou cabdal i, en aquest sentit, la química agrícola il•luminava noves estratègies per reposar els nutrients del sòl. A partir dels anys 1840, el guano, els dipòsits d’excrements d’aus marines presents especialment al litoral pacífic de l’Amèrica del Sud, fou explotat massivament per l’aport de N i de P que oferia. L’explotació del guano superava amb escreix la taxa de reposició i en les darreres dècades del segle XIX hom cercà noves alternatives. En el cas del N, el N2 atmosfèric apareixia com un reservori, i el procés de Haber el va fer assequible a escala industrial a partir dels anys 1910. Però en el cas del P, no hi ha reservori atmosfèric; únicament en condicions molt reductores, hi ha una certa formació de PH3, molt poc rellevant quantitativament. El desenvolupament del forn elèctric, a partir dels anys 1890, possibilità l’explotació directa de l’apatita per obtindre’n fosfats.

Evolució de l’extracció mundial de roca de fosfats
En el 2011, l’extracció de roca fosfàtica fou de 190 milions de tones mètriques. La major part de la roca fosfàtica es destina a l’obtenció d’àcid fosfòric per a la confecció de fertilitzants. Per a obtindre’l se sotmet a la roca a atac amb àcid sulfúric. Alternativament, la roca pot ser atacada amb fòsfor blanc, obtenint-ne àcid fosfòric de major puresa, destinat a la confecció de detergents i d’altres aplicacions. Una part relativament menor es destina a la producció de fòsfor elemental (910 mil tones anuals).
La roca fosfàtica conté apatita i altres molts minerals. Això explica, per exemple, casos com els fangs radioactius que Ercros va acumular per l’activitat de la seva planta de fosfats a Flix: la radioactivitat és deguda a les quantitats de minerals d’urani presents en els fosfats minerals procedents del Sàhara Occidental. L’explotació de la fluoroapatita de Florida als Estats Units comporta com a producte secundari unes quantitats de fluor que es destinen a la fluoridació de l’aigua. Un altre subproducte, l’escòria silícia, es destina a la construcció.

Les reserves de fosfats són un element estratègic per al Sàhara Occidental. Disputat territorialment entre el Regne del Marroc i la República Àrab Saharaui Democràtica, bona part de la història recent d’aquest territori gira al voltant de l’explotació dels fosfats. En la imatge veiem l’evolució de la mina de Bou Craa, entre 1987 (a sota) i el 2000 (a dalt).
D’acord amb la tecnologia extractiva actual, les reserves mundial de roca fosfàtica d’explotació rendible són de 71.000 milions de tones. La demanda de fosfats es troba associada al desenvolupament agrícola. De fet, l’agricultura moderna depèn críticament de fertilitzants rics en N, P i K. La demanda de fosfats creix a un ritme dues vegades superior al creixement de la població humana.
Com en el cas del guano, les reserves de roca fosfàtica no es reposen al ritme que són explotades. Per analogia amb el concepte de “pic” que M. K. Hubbert va enunciar en el 1956 per al petroli, hom parla d’un “pic del fòsfor”. La idea és que la demanda creixent de fosfats provoca l’esgotament d’aquelles reserves de roca fosfàtica més accessibles. Ja hem vist abans com, al capdavall, el 0,1% de l’escorça terrestre és feta de fòsfor. No obstant, les tecnologies actuals (i les previsibles en un futur proper) tan sols poden treballar amb roques fosfàtiques que tinguin una concentració mínima d’apatites.
Així el pic de fòsfor podria tindre lloc en qüestió de 30 anys. A partir de llavors, el preu dels fosfats es dispararia, amenaçant la producció agrícola mundial. Si atenem al parer de l’International Fertilizer Development Center, aquesta és una estimació alarmant, i les reserves de fosfats podrien resistir durant més de tres segles.
Sigui com sigui, és creixent l’interès per un millor control del cicle del fòsfor. El reciclatge dels fosfats en els ecosistemes més humanitzats esdevé cabdal per reduir la dependència envers els fertilitzants.
Per fer servir l’expressió popularitzada recentment per John Bellamy Foster, la clivella metabòlica (“metabòlic rift”) entre la humanitat i la resta de la natura es pot remeiar amb una millor gestió dels residus urbans. Els macronutrients agrícoles bàsics són el N, el P i el K. El 94% dels compostos NPK que elimina el cos humà, ho són per l’orina. Ara bé, la utilització de l’orina humana com a fertilitzant és limitada per les concentracions de metalls pesats (Pb, Hg, Cd), per la salinitat (degut particularment a les concentracions de Na+ i Cl– i pel fet que la ratio N:P és massa elevada. En tot cas, es calcula que el 70% del N dels residus urbans i el 50% del P i del K, procedeixen de la orina humana (quan, l’orina humana, en terme de volum suposa tan sols un 1% dels residus urbans).

Inodor dissenyat per a una recollida separada de l’orina a Johannesburg. La riquesa en NPK de l’orina humana ha fet que mesures de separació de l’orina s’hagin implantat en països com Sud-Àfrica, Xina o Suècia. La gestió de l’orina permet d’una banda reduir els nivells de NPK dels residus urbans i, en conseqüència, els problemes d’eutrofització en els llocs d’abocament. De l’altra banda, adequadament gestionat, és una font de NPK per a la fertilització de sòls agrícoles
Un altre exemple de mesura en pro del reciclatge de macronutrients és la gestió de l’estruvita que es forma en instal•lacions de depuradores d’aigües residuals. L’estruvita (NH4MgPO4•6H2O) es forma en aigües residuals on les concentració de magnesi (Mg2+), amoni (NH4+) i fosfat (PO43+) assoleixin nivells similars. Els cristalls d’estruvita poden obturar aquests sistemes, de manera que un mecanisme d’eliminació que recuperi el material i el destini a la confecció de fertilitzants esdevé interessant.

{kind=link}
{kind=link}
{kind=link}